1. NÁZEV PŘÍPRAVKU
Cosentyx 150 mg injekční roztok v předplněné injekční stříkačce
Cosentyx 300 mg injekční roztok v předplněné injekční stříkačce
Cosentyx 150 mg injekční roztok v předplněném peru
Cosentyx 300 mg injekční roztok v předplněném peru
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Cosentyx 150 mg injekční roztok v předplněné injekční stříkačce
Jedna předplněná injekční stříkačka obsahuje sekukinumabum 150 mg v 1 ml.
Cosentyx 300 mg injekční roztok v předplněné injekční stříkačce
Jedna předplněná injekční stříkačka obsahuje sekukinumabum 300 mg ve 2 ml.
Cosentyx 150 mg injekční roztok v předplněném peru
Jedno předplněné pero obsahuje sekukinumabum 150 mg v 1 ml.
Cosentyx 300 mg injekční roztok v předplněném peru
Jedno předplněné pero obsahuje sekukinumabum 300 mg ve 2 ml.
Sekukinumab je rekombinantní plně humánní monoklonální protilátka produkovaná v buňkách ovarií čínského křečíka (CHO).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok (injekce)
Roztok je čirý a bezbarvý až mírně nažloutlý.
4. KLINICKÉ ÚDAJE
5. FARMAKOLOGICKÉ VLASTNOSTI
-
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresiva, inhibitory interleukinu, ATC kód: L04AC10
-
Klinická účinnost a bezpečnost
Ložisková psoriáza dospělých
Bezpečnost a účinnost sekukinumabu byly hodnoceny ve čtyřech randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích fáze III u pacientů se středně závažnou až závažnou ložiskovou psoriázou, kteří byli kandidáty pro fototerapii nebo systémovou terapii [ERASURE, FIXTURE, FEATURE, JUNCTURE]. Účinnost a bezpečnost 150 mg a 300 mg sekukinumabu byla hodnocena buď proti placebu nebo proti etanerceptu. Navíc jedna studie hodnotila chronický léčebný režim v porovnání s „přeléčením podle potřeby“ [SCULPTURE].Z 2 403 pacientů, kteří byli zařazeni do placebem kontrolovaných studií bylo 79 % biologicky naivních, 45 % bylo nebiologických selhání a 8 % byla biologická selhání (6 % anti‑TNF selhání a 2 % anti‑p40 selhání). Přibližně 15 až 25 % pacientů ve studiích fáze III mělo při zahájení léčby psoriatickou artritidu (PsA).
Psoriatická studie 1 (ERASURE) hodnotila 738 pacientů. Pacienti randomizovaní na sekukinumab dostávali dávku 150 mg nebo 300 mg v týdnech 0, 1, 2, 3 a 4, následované stejnou dávkou každý měsíc. Psoriatická studie 2 (FIXTURE) hodnotila 1 306 pacientů. Pacienti randomizovaní na sekukinumab dostávali dávku 150 mg nebo 300 mg v týdnech 0, 1, 2, 3 a 4, následované stejnou dávkou každý měsíc. Pacienti randomizovaní na etanercept dostávali dávku 50 mg dvakrát týdně po dobu 12 týdnů, následované dávkou 50 mg každý týden. V obou studiích 1 a 2 byli pacienti randomizovaní na placebo a neodpovídající na léčbu v týdnu 12 převedeni na sekukinumab (buď 150 mg nebo 300 mg) v týdnech 12, 13, 14, a 15, následované stejnou dávkou každý měsíc počínaje týdnem 16. Všichni pacienti byli sledováni po dobu až 52 týdnů od prvního podání medikace ve studii.
Psoriatická studie 3 (FEATURE) hodnotila 177 pacientů používajících předplněné injekční stříkačky v porovnání s placebem po 12 týdnech léčby pro zhodnocení bezpečnosti, tolerability a použitelnosti samoaplikace sekukinumabu pomocí předplněné injekční stříkačky. Psoriatická studie 4 (JUNCTURE) hodnotila 182 pacientů používajících předplněné pero v porovnání s placebem po 12 týdnech léčby pro zhodnocení bezpečnosti, snášenlivosti a použitelnosti samoaplikace sekukinumab pomocí předplněného pera. V obou studiích 3 a 4 pacienti randomizovaní na sekukinumab dostávali dávku 150 mg nebo 300 mg v týdnech 0, 1, 2, 3 a 4, následované stejnou dávkou každý měsíc. Pacienti byli též randomizováni na placebo v týdnech 0, 1, 2, 3 a 4, následované stejnou dávkou každý měsíc.
Psoriatická studie 5 (SCULPTURE) hodnotila 966 pacientů. Všichni pacienti dostávali sekukinumab v dávce 150 mg nebo 300 mg v týdnech 0, 1, 2, 3, 4, 8 a 12 a poté byli randomizováni buď k udržovací léčbě stejnou dávkou každý měsíc počínaje týdnem 12, nebo na režim s „přeléčením v čas potřeby“ stejnou dávkou. U pacientů randomizovaných k „přeléčení v čas potřeby“ nebylo dosaženo odpovídající udržení odpovědi a proto se doporučuje fixní měsíční udržovací režim.
Ko-primární cíl placebem a aktivním komparátorem kontrolovaných studií činil podíl pacientů s dosaženou odpovědí PASI 75 a IGA mód 2011 odpovědi “čistý” nebo “téměř čistý” v porovnání s placebem v týdnu 12 (viz tabulky 4 a 5). Dávka 300 mg poskytovala zlepšenou clearance pokožky zejména pro “čistou” nebo “téměř čistou” pokožku v rozmezí cílů účinnosti PASI 90, PASI 100, a IGA mód 2011 0 nebo 1 odpověď u všech studií s největším účinkem pozorovaným v týdnu 16, proto se doporučuje tato dávka.
Tabulka 4 Souhrnná PASI 50/75/90/100 & IGA* mód 2011 “čistá” nebo “téměř čistá” klinická odpověď v psoriatických studiích 1, 3 a 4 (ERASURE, FEATURE a JUNCTURE)
Týden 12
Týden 16
Týden 52
Placebo
150 mg
300 mg
150 mg
300 mg
150 mg
300 mg
Studie 1
Počet pacientů
246
244
245
244
245
244
245
Odpověď PASI 50 n (%)
22 (8,9 %)
203 (83,5 %)
222 (90,6 %)
212 (87,2 %)
224 (91,4 %)
187 (77 %)
207 (84,5 %)
Odpověď PASI 75 n (%)
11 (4,5 %)
174 (71,6 %)**
200 (81,6 %)**
188 (77,4 %)
211 (86,1 %)
146 (60,1 %)
182 (74,3 %)
Odpověď PASI 90 n (%)
3 (1,2 %)
95 (39,1 %)**
145 (59,2 %)**
130 (53,5 %)
171 (69,8 %)
88 (36,2 %)
147 (60,0 %)
Odpověď PASI 100 Odpověď n (%)
2 (0,8 %)
31 (12,8 %)
70 (28,6 %)
51 (21,0 %)
102 (41,6 %)
49 (20,2 %)
96 (39,2 %)
Odpověď IGA mód 2011 “čistý” nebo “téměř čistý” n (%)
6 (2,40 %)
125 (51,2 %)**
160 (65,3 %)**
142 (58,2 %)
180 (73,5 %)
101 (41,4 %)
148 (60,4 %)
Studie 3
Počet pacientů
59
59
58
‑
‑
‑
‑
Odpověď PASI 50 n (%)
3 (5,1 %)
51 (86,4 %)
51 (87,9 %)
‑
‑
‑
‑
Odpověď PASI 75 n (%)
0 (0,0 %)
41 (69,5 %)**
44 (75,9 %)**
‑
‑
‑
‑
Odpověď PASI 90 n (%)
0 (0,0 %)
27 (45,8 %)
35 (60,3 %)
‑
‑
‑
‑
Odpověď PASI 100 n (%)
0 (0,0 %)
5
(8,5 %)25 (43,1 %)
‑
‑
‑
‑
Odpověď IGA mód 2011 “čistý” nebo “téměř čistý” n (%)
0 (0,0 %)
31 (52,5 %)**
40 (69,0 %)**
‑
‑
‑
‑
Studie 4
Počet pacientů
61
60
60
‑
‑
‑
‑
Odpověď PASI 50 n (%)
5 (8,2 %)
48 (80,0 %)
58 (96,7 %)
‑
‑
‑
‑
Odpověď PASI 75 n (%)
2 (3,3 %)
43 (71,7 %)**
52 (86,7 %)**
‑
‑
‑
‑
Odpověď PASI 90 n (%)
0 (0,0 %)
24 (40,0 %)
33 (55,0 %)
‑
‑
‑
‑
Odpověď PASI 100 n (%)
0 (0,0 %)
10 (16,7 %)
16 (26,7 %)
‑
‑
‑
‑
Odpověď IGA mód 2011 “čistý” nebo “téměř čistý” n (%)
0 (0,0 %)
32 (53,3 %)**
44 (73,3 %)**
‑
‑
‑
‑
* IGA mód 2011 je 5 bodová škála zahrnující “0 = čistý”, “1 = téměř čistý”, “2 = mírný”, “3 = středně závažný” nebo “4 = závažný”, indikující lékařovo celkové hodnocení závažnosti psoriázy zaměřené na ztvrdnutí, erytém a odlupování. Léčebný úspěch “ čistý ” nebo “ téměř čistý ” znamenaly nepřítomnost známek psoriázy nebo normální až růžové zbarvené léze, netloustnutí ložisek a žádné nebo minimální místní odlupování.
** p-hodnoty versus placebo a nastavené na multiplicitu: p<0,0001.
Tabulka 5 Souhrn klinické odpovědi v psoriatické studii 2 (FIXTURE)
Týden 12
Týden 16
Týden 52
Placebo
150 mg
300 mg
Etanercept
150 mg
300 mg
Etanercept
150 mg
300 mg
Etanercept
Počet pacientů
324
327
323
323
327
323
323
327
323
323
Odpověď PASI 50 n (%)
49 (15,1 %)
266 (81,3 %)
296 (91,6 %)
226 (70,0 %)
290 (88,7 %)
302 (93,5 %)
257 (79,6 %)
249 (76,1 %)
274 (84,8 %)
234 (72,4 %)
Odpověď PASI 75 n (%)
16 (4,9 %)
219 (67,0 %)**
249 (77,1 %)**
142 (44,0 %)
247 (75,5 %)
280 (86,7 %)
189 (58,5 %)
215 (65,7 %)
254 (78,6 %)
179 (55,4 %)
Odpověď PASI 90 n (%)
5 (1,5 %)
137 (41,9 %)
175 (54,2 %)
67 (20,7 %)
176 (53,8 %)
234 (72,4 %)
101 (31,3 %)
147 (45,0 %)
210 (65,0 %)
108 (33,4 %)
Odpověď PASI 100 n (%)
0 (0 %)
47 (14,4 %)
78 (24,1 %)
14 (4,3 %)
84 (25,7 %)
119 (36,8 %)
24 (7,4 %)
65 (19,9 %)
117 (36,2 %)
32 (9,9 %)
Odpověď IGA mód 2011 “čistý” nebo “téměř čistý” n (%)
9 (2,8 %)
167 (51,1 %)**
202 (62,5 %)**
88 (27,2 %)
200 (61,2 %)
244 (75,5 %)
127 (39,3 %)
168 (51,4 %)
219 (67,8 %)
120 (37,2 %)
** p-hodnoty versus etanercept: p=0,0250
V další psoriatické studii (CLEAR) bylo hodnoceno 676 pacientů. V této studii byla s ohledem na primární a sekundární cílové parametry prokázána superiorita sekukinumabu v dávce 300 mg oproti ustekinumabu v odpovědích PASI 90 v 16. týdnu (primární cílový parametr), v rychlosti nástupu odpovědi PASI 75 ve 4. týdnu a v dlouhodobé odpovědi PASI 90 v 52. týdnu. Vyšší účinnost sekukinumabu v porovnání s ustekinumabem pro cílové parametry PASI 75/90/100 a odpověď IGA mod 2011 0 nebo 1 („čistý“ nebo „téměř čistý“) byla pozorována po celou dobu, od začátku do 52. týdne (tabulka 6).
Tabulka 6 Souhrn klinické odpovědi ve studii CLEAR
Týden 4
Týden 16
Týden 52
Sekukinumab 300 mg
Ustekinumab*
Sekukinumab 300 mg
Ustekinumab*
Sekukinumab 300 mg
Ustekinumab*
Počet pacientů
334
335
334
335
334
335
Odpověď PASI 75 n (%)
166 (49,7 %)**
69 (20,6 %)
311 (93,1 %)
276 (82,4 %)
306 (91,6 %)
262 (78,2 %)
Odpověď PASI 90 n (%)
70 (21,0 %)
18 (5,4 %)
264 (79,0 %)**
192 (57,3 %)
250 (74,9 %)***
203 (60,6 %)
Odpověď PASI 100 n (%)
14 (4,2 %)
3 (0,9 %)
148 (44,3 %)
95 (28,4 %)
150 (44,9 %)
123 (36,7 %)
Odpověď IGA mód 2011 „čistý“ nebo „téměř čistý“ n (%)
128 (38,3 %)
41 (12,2 %)
278 (83,2 %)
226 (67,5 %)
261 (78,1 %)
213 (63,6 %)
* Pacienti léčení sekukinumabem dostávali dávku 300 mg v týdnech 0, 1, 2, 3 a 4 následovanou stejnou dávkou vždy po 4 týdnech do 52. týdne. Pacienti léčení ustekinumabem dostávali dávku 45 mg nebo 90 mg v týdnech 0 a 4, a poté vždy po 12 týdnech do 52. týdne (dávka podle hmotnosti a schváleného dávkování)
** p-hodnoty versus ustekinumab: p<0,0001 pro primární cílový parametr PASI90 v 16. týdnu a sekundární cílový parametr PASI75 ve 4. týdnu
*** p-hodnoty versus ustekinumab: p<0,0001 pro sekundární cílový parametr PASI90 v 52. týdnuSekukinumab byl účinný u pacientů bez předchozí systémové léčby, biologicky naivních, biologicky/anti‑TNF‑exponovaných a pacientů s biologickým/anti‑TNF selháním. Zlepšení PASI 75 u pacientů se souběžnou psoriatickou artritidou při zahájení léčby byly podobné těm u celé populace s ložiskovou psoriázou.
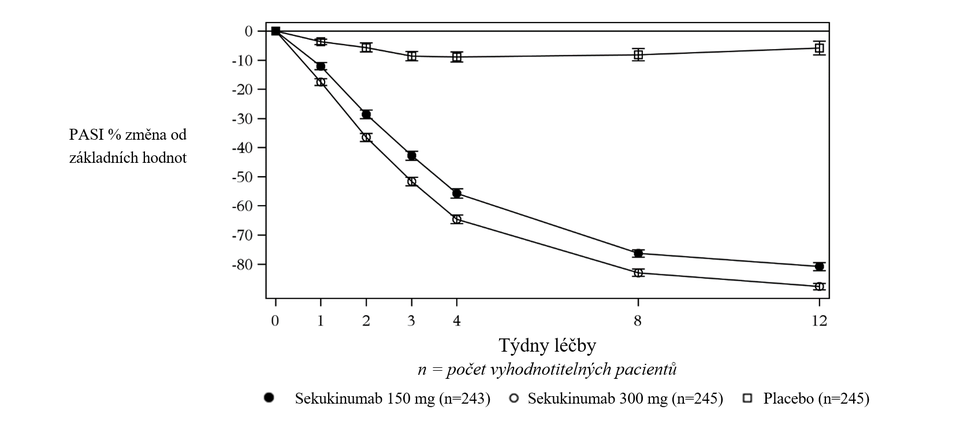
Sekukinumab byl spojován s rychlým nástupem účinku s 50% redukcí průměrného PASI do týdne 3 při dávce 300 mg.
Obrázek 1 Časový průběh procentní změny od výchozích hodnot průměrného PASI skóre ve studii 1 (ERASURE)
Specifické lokalizace/formy ložiskové psoriázy
Ve dvou dalších placebem kontrolovaných studiích bylo pozorováno zlepšení jak u nehtové psoriázy (studie TRANSFIGURE, 198 pacientů), tak u palmoplantární ložiskové psoriázy (studie GESTURE, 205 pacientů). Ve studii TRANSFIGURE byla prokázána superiorita sekukinumabu oproti placebu v 16. týdnu (46,1 % pro 300 mg, 38,4 % pro 150 mg a 11,7 % pro placebo) ve výrazném zlepšení oproti počátečnímu stavu podle indexu závažnosti psoriázy nehtů (NAPSI %) u pacientů se středně těžkou až těžkou ložiskovou psoriázou s postižením nehtů. Ve studii GESTURE byla prokázána superiorita sekukinumabu oproti placebu v 16. týdnu (33,3 % pro 300 mg, 22,1 % pro 150 mg a 1,5 % pro placebo) ve výrazném zlepšení oproti počátečnímu stavu podle odpovědi ppIGA 0 nebo 1 („čistý“ nebo „téměř čistý“) u pacientů se středně těžkou až těžkou palmoplantární ložiskovou psoriázou.V placebem kontrolované studii bylo hodnoceno 102 pacientů se středně těžkou až těžkou psoriázou ve vlasové části hlavy, definovanou hodnotou Psoriasis Scalp Severity Index (PSSI) ≥12, odpovědí dle skóre IGA, mod 2011, pouze ve vlasové části hlavy skóre 3 nebo větší a postižením nejméně 30 % plochy vlasové části. Ve 12. týdnu byla prokázána superiorita sekukinumabu 300 mg v porovnání s placebem ve výrazném zlepšení oproti počátečnímu stavu jak pro odpovědi PSSI 90 (52,9 % versus 2,0 %), tak u odpovědí dle skóre IGA, mod 2011, 0 nebo 1 pouze ve vlasové části hlavy (56,9 % versus 5,9 %). U pacientů pokračujících v léčbě sekukinumabem do 24. týdne bylo zlepšení hodnocené dle obou cílových parametrů setrvalé.
Kvalita života/výsledky hlášené pacienty
Statisticky zlepšení hodnot v týdnu 12 (studie 1‑4) oproti hodnotám před léčbou v porovnání s placebem bylo demonstrováno pomocí DLQI (Dermatology Life Quality Index). Průměrný pokles (zlepšení) DLQI z hodnot před léčbou kolísalo od ‑10,4 do ‑11,6 u sekukinumabu 300 mg, od ‑7,7 do ‑10,1 u sekukinumabu 150 mg, v porovnání s ‑1,1 až ‑1,9 u placeba v týdnu 12. Toto zlepšení přetrvávalo 52 týdnů (studie 1 a 2).Čtyřicet procent účastníků ve studiích 1 a 2 vyplnilo deník Psoriasis Symptom Diary©. U účastníků, kteří vyplnili tento deník, bylo v každé z těchto studií v porovnání s placebem prokázáno statisticky významné zlepšení z hodnot před léčbou v týdnu 12 u pacienty hlášených známek a příznaků svědění, bolesti a odlupování.
U pacientů léčených sekukinumabem, v porovnání s pacienty léčenými ustekinumabem (CLEAR), bylo ve 4. týdnu dle hodnoty DLQI prokázáno statisticky významné zlepšení oproti výchozímu stavu. Toto zlepšení přetrvávalo až do 52. týdne.
Záznamy v deníku Psoriasis Symptom Diary© doložily v 16. a 52. týdnu u pacientů léčených sekukinumabem, v porovnání s pacienty léčenými ustekinumabem, statisticky významné zlepšení v hodnocení pacienty hlášených známek a příznaků svědění, bolesti a odlupování (CLEAR).
Ve studii s pacienty s psoriázou ve vlasové části hlavy bylo ve 12. týdnu, v porovnání s placebem, prokázáno statisticky významné zlepšení oproti výchozímu stavu (pokles) v hodnocení známek a příznaků svědění, bolesti a odlupování hlášených pacienty.
Flexibilita dávky u ložiskové psoriázy
Randomizovaná, dvojitě zaslepená, multicentrická studie hodnotila dva udržovací dávkovací režimy (300 mg každé 2 týdny a 300 mg každé 4 týdny), dávky byly podávané 150 mg předplněnou injekční stříkačkou u 331 pacientů vážících ≥90 kg se středně těžkou až těžkou psoriázou.
Pacienti byli randomizováni v poměru 1:1 následovně:- sekukinumab 300 mg v týdnech 0, 1, 2, 3 a 4 následovaný stejnou dávkou každé 2 týdny
až do týdne 52 (n=165). - sekukinumab 300 mg v týdnech 0, 1, 2, 3 a 4 následovaný stejnou dávkou každé 4 týdny
až do týdne 16 (n=166).
-
- Pacienti randomizovaní k léčbě sekukinumabem 300 mg každé 4 týdny, kteří v týdnu 16 reagovali odpovědí PASI 90, pokračovali v podávání stejného dávkovacího režimu až do týdne 52.
Pacienti randomizovaní k léčbě sekukinumabem 300 mg každé 4 týdny, kteří v týdnu 16 nedosáhli odpovědi PASI 90, buď pokračovali v stejném dávkovacím režimu nebo byli přeřazeni na léčbu sekukinumabem 300 mg každé 2 týdny až do týdne 52.
- Pacienti randomizovaní k léčbě sekukinumabem 300 mg každé 4 týdny, kteří v týdnu 16 reagovali odpovědí PASI 90, pokračovali v podávání stejného dávkovacího režimu až do týdne 52.
Celkově byla míra odpovědi na léčbu vyšší u skupiny léčené v režimu každé 2 týdny v porovnání se
skupinou léčenou v režimu každé 4 týdny (tabulka 7).Tabulka 7 Shrnutí klinické odpovědi ve studii s flexibilitou dávky u ložiskové psoriázy*
Týden 16
Týden 52
sekukinumab
300 mg Q2Wsekukinumab
300 mg Q4Wsekukinumab
300 mg Q2Wsekukinumab
300 mg Q4W1Počet pacientů
165
166
165
83
Odpověď PASI 90 n (%)
121 (73,2%) **
92 (55,5%)
126 (76,4%)
44 (52,4%)
IGA mód 2011 “čistý” nebo “téměř čistý” n (%)
122 (74,2%)2
109 (65,9%)2
125 (75,9%)
46 (55,6%)
* Mnohočetné imputace
1 300 mg Q4W: pacienti kontinuálně léčení 300 mg Q4W bez ohledu na stav odpovědi PASI 90 v 16. týdnu;
43 pacientů bylo PASI 90 respondérů v týdnu 16 a 40 pacientů bylo PASI 90 non-respondérů v týdnu 16
** Jednostranná hodnota p = 0,0003 pro primární cílový parametr PASI 90 v týdnu 16
2 Hodnota není statisticky významná
Q2W: každé 2 týdny; Q4W: každé 4 týdnyU pacientů, kteří nereagovali odpovědí PASI 90 v 16. týdnu, u kterých byla dávka titrována na sekukinumab 300 mg každé 2 týdny, se míra odpovědi PASI 90 zlepšila v porovnání s těmi, kteří zůstali na dávkovacím režimu sekukinumab 300 mg každé 4 týdny, zatímco míra odpovědí IGA mód 2011 0/1 zůstala v průběhu času stabilní u obou léčebných skupin.
Bezpečnostní profily dvou dávkovacích režimů, Cosentyx 300 mg podávaný každé 4 týdny a Cosentyx 300 mg podávaný každé 2 týdny, u pacientů s hmotností ≥90 kg byly srovnatelné a konzistentní s bezpečnostním profilem hlášeným u pacientů s psoriázou.
Hidradenitida
Bezpečnost a účinnost sekukinumabu byly hodnoceny u 1 084 pacientů ve dvou randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích fáze III u dospělých pacientů se středně těžkou až těžkou hidradenitidou (HS), kteří byli kandidáti pro systémovou biologickou léčbu. Na počátku léčby měli pacienti nejméně pět zánětlivých lézí postihujících alespoň dvě anatomické oblasti. V HS studii 1 (SUNSHINE) bylo I. stupněm klasifikováno 4,6 % pacientů , II. stupněm 61,4 % pacientů a III. stupněm Hurleyho skóre 34 % pacientů. V HS studii 2 (SUNRISE) bylo I. stupněm klasifikováno 2,8 % pacientů , II. stupněm 56,7 % pacientů a III. stupněm Hurleyho skóre 40,5 % pacientů. Poměr pacientů s hmotností ≥90 kg byl 54,7 % v HS studii 1 a 50,8 % v HS studii 2. Pacientům v těchto studiích byla průměrně před 7,3 lety diagnostikována středně těžká až těžká HS a 56,3 % účastníků studie byly ženy.
V HS studii 1 bylo 23,8 % pacientů v minulosti léčeno biologickou léčbou a celkem 82,3 % pacientů bylo v minulosti léčeno systémovými antibiotiky. V HS studii 2 bylo 23,2 % pacientů v minulosti léčeno biologickou léčbou a celkem 83,6 % pacientů bylo v minulosti léčeno systémovými antibiotiky.
V HS studii 1 bylo hodnoceno 541 pacientů, v HS studii 2 bylo hodnoceno 543 pacientů, z nichž 12,8 % resp. 10,7 % souběžně užívalo stálou dávku antibiotik. V obou studiích byli pacienti randomizováni na 300 mg sekukinumabu subkutánně v týdnech 0, 1, 2, 3 a 4 a dále 300 mg každé 2 týdny nebo každé 4 týdny. Pacienti, kteří byli randomizováni na placebo, byli v 16. týdnu převedeni na sekukinumab 300 mg v týdnech 16, 17, 18, 19 a 20 a dále sekukinumab 300 mg každé 2 týdny nebo sekukinumab 300 mg každé 4 týdny.
Primární cílový parametr v obou studiích (HS studii 1 a HS studii 2) byl poměr pacientů, kteří v 16. týdnu dosáhli klinické odpovědi definované jako alespoň 50% snížení počtu abscesů a zánětlivých nodulů bez zvýšení množství abscesů a/nebo množství drénujících fistul vzhledem k výchozí hodnotě (HiSCR50). Snížení bolesti kůže související s HS bylo hodnoceno jako sekundární cílový parametr na základě souhrnných údajů z HS studie 1 a HS studie 2 získaných pomocí číselné hodnoticí škály (NRS, Numerical Rating Scale) od pacientů, kteří vstoupili do studie s počátečním skóre 3 nebo vyšším.
V HS studii 1 a HS studii 2 dosáhl v 16. týdnu vyšší podíl pacientů léčených sekukinumabem 300 mg každé 2 týdny odpovědi HiSCR50 s poklesem počtu abscesů a zánětlivých nodulů (AN) ve srovnání s placebem. V HS studii 2 byl rovněž pozorován rozdíl v odpovědi HiSCR a počtu AN ve skupině se sekukinumabem 300 mg každé 4 týdny. Ve skupině se sekukinumabem 300 mg každé 2 týdny v HS studii 1 a ve skupině se sekukinumabem 300 mg každé 4 týdny v HS studii 2 došlo ve srovnání s placebem ke vzplanutí onemocnění do 16. týdne s nižší četností. Vyšší poměr pacientů léčených sekukinumabem 300 mg každé 2 týdny (souhrnné údaje) zaznamenal do 16. týdne klinicky významný pokles bolesti kůže související s HS ve srovnání s placebem (tabulka 8).
Tabulka 8 Klinická odpověď v HS studii 1 a HS studii 2 v 16. týdnu1
HS studie 1 HS studie 2 Placebo 300 mg Q4W 300 mg Q2W Placebo 300 mg Q4W 300 mg Q2W Počet randomizovaných pacientů 180 180 181 183 180 180 HiSCR50, n (%) 61
(33,7)75
(41,8)82
(45,0*)57
(31,2)83
(46,1*)76
(42,3*)Počet AN, průměrná změna oproti výchozí hodnotě, % ‑24,3 ‑42,4 ‑46,8* ‑22,4 ‑45,5* ‑39,3* Vzplanutí, n (%) 52
(29,0)42
(23,2)28
(15,4*)50
(27,0)28
(15,6*)36
(20,1)Souhrnné údaje (HS studie 1 a HS studie 2) Placebo 300 mg Q4W 300 mg Q2W Počet pacientů s výchozí hodnotou NRS ≥3 251 252 266 ≥30% redukce bolesti kůže, NRS30 odpověď, n (%) 58 (23,0) 84 (33,5) 97 (36,6*) 1 Ke zpracování chybějících údajů byla použita vícenásobná imputace
n: Zaokrouhlený průměr počtu subjektů s odpověďmi ve 100 imputacích
* Statisticky významné v porovnání s placebem na základě predefinované hierarchie s celkovou hodnotou ɑ=0,05
AN: abscesy a zánětlivé noduly; HiSCR: klinická odpověď hidradenitidy; NRS: číselná hodnoticí škála; Q4W: každé 4 týdny; Q2W: každé 2 týdnyV obou studiích se účinek sekukinumabu projevil již ve 2. týdnu, postupně rostl do 16. týdne a přetrval až do 52. týdne.
U primárního a nejdůležitějšího sekundárního cílového parametru bylo pozorováno zlepšení u pacientů s HS nezávisle na předchozí nebo souběžné antibiotické léčbě.
Zlepšení v parametru HiSCR50 bylo v 16. týdnu dosaženo jak u pacientů bez předchozí biologické léčby, tak u pacientů již vystavených biologické léčbě.
Výraznější zlepšení oproti výchozí hodnotě ve srovnání s placebem bylo prokázáno v 16. týdnu v kvalitě života související se zdravím podle dermatologického indexu kvality života (DLQI, Dermatology Life Quality Index).
Psoriatická artritida
Bezpečnost a účinnost sekukinumabu byla hodnocena u 1 999 pacientů ve třech randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích fáze III u pacientů s aktivní psoriatickou artritidou (≥3 oteklé a ≥3 bolestivé klouby) přes léčbu nesteroidními protizánětlivými léky (NSAID), kortikosteroidy nebo chorobu modifikujícími antirevmatiky (DMARD). Do těchto studií byli zařazeni pacienti všech subtypů PsA, včetně polyartikulární artritidy bez průkazu revmatoidních nodulů, spondylitidy s periferní artritidou, asymetrickou periferní artritidou, postižení distálních interfalangeálních kloubů a arthritis mutilans. Pacienti v těchto studiích měli stanovenu diagnózu PsA nejméně 5 let. Většina pacientů měla též aktivní psoriatické kožní léze nebo dokumentovanou psoriázu v anamnéze. Přes 61 % a 42 % pacientů s PsA mělo před zahájením entezitidu, respektive daktylitidu. Ve všech studiích bylo dosaženo primárního cíle – odpovědi ACR20 (American College of Rheumatology). V 1. studii s psoriatickou artritidou (PsA studie 1) a ve 2. studii s psoriatickou artritidou (PsA studie 2) byl primární cílový parametr ve 24. týdnu. Ve 3. studii s psoriatickou artritidou (PsA studie 3) byl primární cílový parametr v 16. týdnu s nejdůležitějším sekundárním cílovým parametrem, změnou od výchozích hodnot v modifikovaném mTSS (Total Sharp Score), který byl ve 24. týdnu.V PsA studii 1, PsA studii 2 a PsA studii 3 bylo 29 % pacientů, 35 % pacientů a 30 % pacientů, v uvedeném pořadí, již léčeno anti‑TNFα přípravkem a ukončilo užívání anti-TNFα přípravku buď pro nedostatek účinku nebo nesnášenlivost (anti-TNFα-IR pacienti).
PsA studie 1 (FUTURE 1) hodnotila 606 pacientů, z nichž 60,7 % užívalo současně MTX. Pacienti randomizovaní na sekukinumab dostávali dávku 10 mg/kg intravenózně v týdnech 0, 2 a 4, následovanou buď 75 mg nebo 150 mg subkutánně každý měsíc počínaje týdnem 8. Pacienti randomizovaní na placebo byli převedeni na sekukinumab (buď 75 mg nebo 150 mg subkutánně) s následnou stejnou dávkou každý měsíc, non-respondéři v 16. týdnu (časná záchrana) a ostatní ve 24. týdnu.
PsA studie 2 (FUTURE 2) hodnotila 397 pacientů, z nichž 46,6 % užívalo současně MTX. Pacienti randomizovaní na sekukinumab dostávali dávku 75 mg, 150 mg nebo 300 mg subkutánně v týdnech 0, 1, 2, 3 a 4, s následnou stejnou dávkou každý měsíc. Pacienti randomizovaní na placebo, kteří v týdnu 16 neodpovídali na léčbu byli v 16. týdnu (časná záchrana) převedeni na sekukinumab (buď 150 mg nebo 300 mg subkutánně) s následnou stejnou dávkou každý měsíc. Pacienti randomizovaní na placebo, kteří v 16. týdnu měli odpověď na léčbu, byli v 24. týdnu převedeni na sekukinumab (buď 150 mg nebo 300 mg subkutánně) s následnou stejnou dávkou každý měsíc.
PsA studie 3 (FUTURE 5) hodnotila 996 pacientů, z nichž 50,1 % užívalo současně MTX. Pacienti byli randomizováni do ramen se sekukinumabem 150 mg, 300 mg nebo placebo podávaných subkutánně v týdnech 0, 1, 2, 3 a 4, následovala stejná dávka každý měsíc nebo injekce obsahující 150 mg sekukinumabu aplikovaná jednou měsíčně (bez zaváděcích dávek). Pacienti randomizovaní do ramene s placebem, kteří neodpovídali na léčbu v týdnu 16 (časná záchrana) byli poté přeřazeni do ramene s podáváním sekukinumabu (buď 150 mg nebo 300 mg subkutánně) v týdnu 16, následovaným stejnou dávkou každý měsíc. Pacienti randomizovaní k užívání placeba, kteří odpovídali na léčbu v týdnu 16, byli přeřazeni do ramene s užíváním sekukinumabu (buď 150 mg nebo 300 mg subkutánně) v týdnu 24, následovaným stejnou dávkou každý měsíc.
Známky a příznaky
Léčba sekukinumabem v porovnání s placebem znamenala významné zlepšení v hodnocení aktivity choroby v týdnech 16 a 24 (viz tabulka 9).Tabulka 9 Klinická odpověď u PsA studie 2 a PsA studie 3 v týdnu 16 a v týdnu 24
PsA studie 2
PsA studie 3
Placebo
150 mg1
300 mg1
Placebo
150 mg1
300 mg1
Počet randomizovaných pacientů
98
100
100
332
220
222
ACR20 odpověď
n (%)týden 16
18
(18,4%)60
(60,0%***)57
(57,0%***)91◊
(27,4%)122◊
(55,5%***)139◊
(62,6%***)týden 24
15◊
(15,3%)51◊
(51,0%***)54◊
(54,0%***)78
(23,5%)117
(53,2%***)141
(63,5%***)ACR50 odpověď
n (%)týden 16
6
(6,1 %)37
(37,0%***)35
(35,0%***)27
(8,1%)79
(35,9%*)88
(39,6%*)týden 24
7
(7,1%)35
(35,0%)35
(35,0%**)29
(8,7%)86
(39,1%***)97
(43,7%***)ACR70 odpověď
n (%)týden 16
2
(2,0%)17
(17,0%**)15
(15,0%**)14
(4,2%)40
(18,2%***)45
(20,3%***)týden 24
1
(1,0%)21
(21,0%**)20
(20,0%**)13
(3,9%)53
(24,1%***)57
(25,7%***)DAS28‑CRP
týden 16
-0,50
-1,45***
-1,51***
-0,63
-1,29*
-1,49*
týden 24
-0,96
-1,58**
-1,61**
-0,84
-1,57***
-1,68***
Počet pacientů s ≥3% BSA psoriatickým postižením pokožky před zahájením léčby
43
(43,9%)58
(58,0%)41
(41,0%)162
(48,8%)125
(56,8%)110
(49,5%)PASI 75 odpověď
n (%)týden 16
3
(7,0%)33
(56,9%***)27
(65,9%***)20
(12,3%)75
(60,0%*)77
(70,0%*)týden 24
7
(16,3%)28
(48,3%**)26
(63,4%***)29
(17,9%)80
(64,0%***)78
(70,9%***)PASI 90 odpověď
n (%)týden 16
3
(7,0%)22
(37,9%***)18
(43,9%***)15
(9,3%)46
(36,8%*)59
(53,6%*)týden 24
4
(9,3%)19
(32,8%**)20
(48,8%***)19
(11,7%)51
(40,8%***)60
(54,5%***)Vymizení daktylitidy n (%) †
týden 16
10
(37%)21
(65,6%*)26
(56,5%)40
(32,3%)46
(57,5%*)54
(65,9%*)týden 24
4
(14,8%)16
(50,0%**)26
(56,5%**)42
(33,9%)51
(63,8%***)52
(63,4%***)Vymizení entezitidy n (%) ‡
týden 16
17
(26,2%)32
(50,0%**)32
(57,1%***)68
(35,4%)77
(54,6%*)78
(55,7%*)týden 24
14
(21,5%)27
(42,2%*)27
(48,2%**)66
(34,4%)77
(54,6%***)86
(61,4%***)* p<0,05, ** p<0,01, *** p<0,001; oproti placebu
Všechny p‑hodnoty jsou nastaveny na mnohočetné testování založené na předem definované hierarchii v týdnu 24 pro PsA studii 2, s výjimkou ACR70, daktylitidy a entezitidy, což byly exploratorní cíle a všechny cíle v týdnu 16.
Všechny p‑hodnoty jsou nastaveny na mnohočetné testování založené na předem definované hierarchii v týdnu 16 pro PsA studii 3, s výjimkou ACR70, což byl exploratorní cíl a všechny cíle v týdnu 24.
Pacienti s chybějícími údaji o léčebné odpovědi byli považováni za non-respondéry.
ACR: American College of Rheumatology; PASI: Psoriasis Area and Severity Index; DAS: Disease Activity Score (skóre aktivity nemoci); BSA: Body Surface Area (povrch těla)
◊Primary Endpoint (primární cíl)
11Sekukinumab 150 mg nebo 300 mg s.c. v týdnech 0, 1, 2, 3 a 4, následovaný stejnou dávkou každý měsíc
†U pacientů s daktylitidou při zahájení (n=27, 32, 46, v uvedeném pořadí pro PsA studii 2 a n=124, 80, 82, v uvedeném pořadí for PsA studii 3)
‡U pacientů s entezitidou při zahájení (n=65, 64, 56, v uvedeném pořadí pro PsA studii 2 a n=192, 141, 140, v uvedeném pořadí pro PsA studii 3)Nástup účinku sekukinumabu byl pozorován již ve 2. týdnu. Statisticky významného rozdílu v ACR20 oproti placebu bylo dosaženo ve 3. týdnu.
Procento pacientů s dosaženou ACR20 odpovědí je na obrázku 2.
Obrázek 2 ACR20 odpověď v PsA studii 2 v závislosti na čase do týdne 52
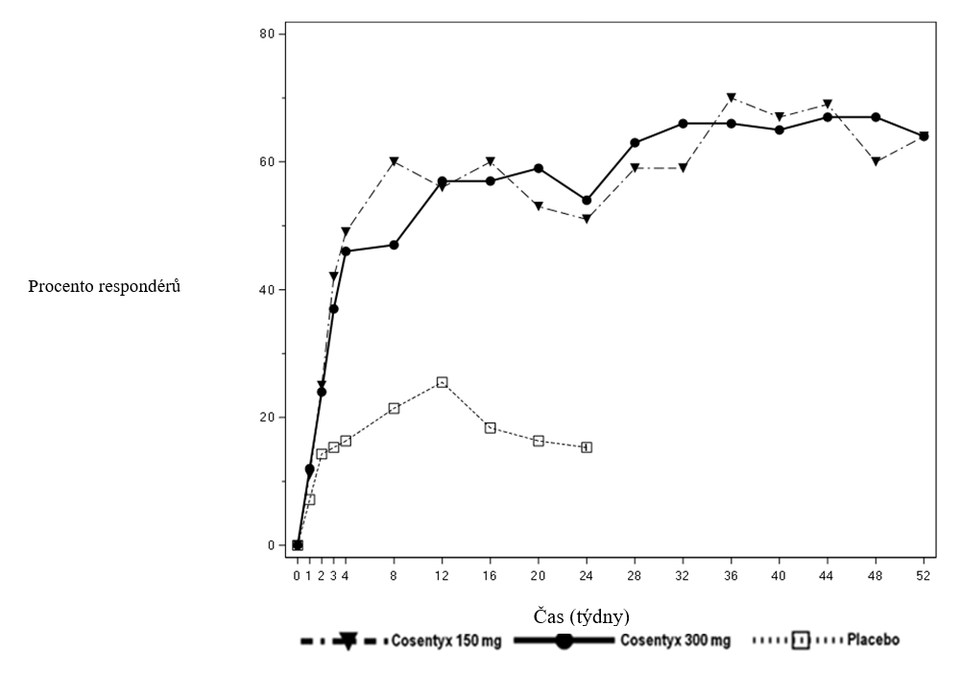
Podobné odpovědi pro primární a klíčový sekundární cílový parametr byly pozorovány u PsA pacientů bez ohledu na to, zda byli současně léčeni MTX či nikoliv. V PsA studii 2 v týdnu 24 měli pacienti léčení sekukinumabem a současně MTX vyšší ACR20 odpověď (47,7 % a 54,4 % u 150 mg a 300 mg, v porovnání s placebem 20,0 %) a ACR 50 odpověď (31,8 % a 38,6 % 150 mg a 300 mg v porovnání s placebem 8,0 %). Pacienti léčení sekukinumabem bez současně podávaného MTX měli vyšší ACR20 odpověď (53,6 % a 53,6 % u 150 mg a 300 mg, v porovnání s placebem 10,4 %) a ACR 50 odpověď (37,5 % a 32,1 % u 150 mg a 300 mg, v porovnání s placebem 6,3 %).
V PsA studii 2 měli jak anti-TNFα naivní, tak anti-TNFα-IR pacienti léčení sekukinumabem významně vyšší ACR20 odpověď v porovnání s placebem v týdnu 24, s mírně vyšší odpovědí u anti-TNFα naivní skupiny (anti-TNFα naivní: 64 % a 58 % u 150 mg a 300 mg, v porovnání s placebem 15,9 %; anti-TNFα-IR: 30 % a 46 % u 150 mg a 300 mg, v porovnání s placebem 14,3 %). V anti-TNFα-IR podskupině pacientů vykázala významně vyšší ACR20 odpověď v porovnání s placebem (p<0,05) pouze dávka 300 mg a demonstrovala klinicky smysluplný prospěch oproti 150 mg také ve více sekundárních cílech. Zlepšení PASI 75 odpovědi bylo pozorováno v obou podskupinách a dávka 300 mg prokázala statisticky signifikantní přínos u anti-TNFα-IR pacientů.
Zlepšení se objevilo u všech komponent ACR skóre, včetně hodnocení bolesti pacientem. Podíl pacientů s dosaženou modifikovanou PsA Response Criteria (PsARC) odpovědí v PsA studii 2 byl větší ve skupině pacientů léčených sekukinumabem (59,0 % a 61,0 % u 150 mg a 300 mg) v porovnání s placebem (26,5 %) v týdnu 24.
V PsA studii 1 a PsA studii 2 přetrvávala účinnost do týdne 104. V PsA studii 2 bylo v 52. týdnu stále léčeno 178 pacientů (89 %) z 200 pacientů iniciálně randomizovaných na sekukinumab 150 mg a 300 mg. Ze 100 pacientů randomizovaných na sekukinumab 150 mg, 64, respektive 39 a 20, vykázalo ACR20/50/70 odpověď. Ze 100 pacientů randomizovaných na sekukinumab 300 mg, 64, respektive 44 a 24 vykázalo ACR20/50/70 odpověď.
Radiografická odpověď
V PsA studii 3 byla inhibice progrese strukturálního poškození hodnocena radiograficky a vyjádřena pomocí modifikovaného mTSS (Total Sharp Score) a jeho komponent, ES (Erosion Score) a JSN (Joint Space Narrowing Score). Radiografické snímky rukou, zápěstí a nohou byly získány při zahájení léčby, v týdnu 16 a/nebo v týdnu 24 a zkontrolovány nezávisle alespoň dvěma hodnotiteli, kterým byly zaslepeny léčebné skupiny a pořadí návštěv. Léčba 150 mg a 300 mg sekukinumabu významně inhibovala míru progrese poškození periferních kloubů v porovnání s léčbou placebem, což bylo změřeno změnou od zahájení v mTSS v týdnu 24 (tabulka 10).Inhibice progrese strukturálního poškození byla také hodnocena v PsA studii 1 v týdnech 24 a 52, v porovnání s výchozími hodnotami. Data z týdne 24 jsou uvedena v tabulce 10.
Tabulka 10 Změna modifikovaného Total Sharp skóre u psoriatické artritidy
PsA studie 3
PsA studie 1
Placebo
n=296Sekukinumab 150 mg1
n=213Sekukinumab 300 mg1
n=217Placebo
n=179Sekukinumab 150 mg2
n=185Celkové skóre
Počáteční hodnoty
(SD)15,0
(38,2)13,5
(25,6)12,9
(23,8)28,4
(63,5)22,3
(48,0)Průměrná změna v týdnu 24
0,50
0,13*
0,02*
0,57
0,13*
*p<0,05 založená na nominální, ale neupravené p-hodnotě
1Sekukinumab 150 mg nebo 300 mg s.c. v týdnech 0, 1, 2, 3 a 4, následovaná stejnou dávkou každý měsíc
210 mg/kg v týdnech 0, 2 a 4, následovaná subkutánními dávkami 75 mg nebo 150 mgV PsA studii 1 přetrvávala inhibice strukturálního poškození při léčbě sekukinumabem do týdne 52.
V PsA studii 3 bylo procento pacientů bez progrese choroby (definované jako změna od výchozích hodnot v mTSS ≤0,5) od randomizace do 24. týdne 80,3 % u sekukinumabu 150 mg, 88,5 % u sekukinumabu 300 mg a 73,6 % u placeba. Účinek inhibice strukturálního poškození byl pozorován u anti-TNFα-naivních a anti-TNFα-IR pacientů a u pacientů léčených současně MTX a bez současného MTX.
V PsA studii 1 bylo procento pacientů bez progrese choroby (definované jako změna od výchozích hodnot mTSS ≤0,5) od randomizace do týdne 24 82,3 % ve větvi se sekukinumabem s dávkovacím schématem 10 mg/kg intravenózního bolusu – 150 mg subkutánně (udržovací dávka) a 75,7 % ve větvi s placebem. Procento pacientů bez progrese choroby od týdne 24 do týdne 52 u sekukinumabu 10 mg/kg ve formě intravenózního bolusu – následovaného 150 mg subkutánně (udržovací dávka) a u pacientů na placebu, kteří byli v týdnu 16 nebo 24 převedeni na 75 mg nebo 150 mg subkutánně (udržovací dávka) každé 4 týdny, bylo 85,7 % a 86,8 %.
Axiální postižení u PsA
Randomizovaná, dvojitě zaslepená, placebem kontrolovaná studie (MAXIMISE) hodnotila účinnost sekukinumabu u 485 pacientů s PsA s axiálním postižením, kteří nebyli dosud léčeni biologickou léčbou a u nichž se nedostavila adekvátní odpověď na předchozí léčbu NSAID. Primární cíl, dosažení minimálně 20% zlepšení dle kritérií Assessment of SpondyloArthritis International Society (ASAS) ve 12. týdnu, byl splněn. Léčba sekukinumabem v dávce 300 mg a 150 mg, ve srovnání s placebem, vedla také k výraznějšímu zmírnění známek a symptomů onemocnění (včetně snížení bolesti páteře oproti výchozímu stavu) a zlepšení fyziologických funkcí (viz tabulka 11).Tabulka 11 Klinická odpověď ve studii MAXIMISE ve 12. týdnu
Placebo
(n=164)150 mg
(n=157)300 mg
(n=164)ASAS 20 odpověď, %
(95% CI)31,2 (24,6, 38,7) 66,3 (58,4, 73,3)* 62,9 (55,2, 70,0)* ASAS 40 odpověď, %
(95% CI)12,2 (7,8, 18,4) 39,5 (32,1, 47,4)** 43,6 (36,2, 51,3)** BASDAI 50, %
(95% CI)9,8 (5,9, 15,6) 32,7 (25,8, 40,5)** 37,4 (30,1, 45,4)** Bolest páteře, VAS
(95% CI)-13,6 (-17,2, -10,0) -28,5 (-32,2, -24,8)** -26,5 (-30,1, -22,9)** Fyziologické funkce, HAQ‑DI
(95% CI)-0,155 (-0,224, -0,086) -0,330 (-0,401,
-0,259)**-0,389 (-0,458,
-0,320)*** p<0,0001; oproti placebu použitím vícenásobné imputace.
** Srovnání s placebem nebylo upraveno pro mnohočetné testování.
ASAS: Assessment of SpondyloArthritis International Society Criteria; BASDAI: Bath Ankylosing Spondylitis Disease Activity Index; VAS: Visual Analog Scale; HAQ‑DI: Health Assessment Questionnaire – Disability Index.Zlepšení dle ASAS 20 a ASAS 40 pro obě dávky sekukinumabu bylo dosaženo ve 4. týdnu a bylo udržováno až 52 týdnů.
Funkční stav a kvalita života
V PsA studii 2 a PsA studii 3, pacienti léčení sekukinumabem 150 mg (p=0,0555 a p<0,0001) a 300 mg (p=0,0040 a p<0,0001) vykázali zlepšení funkčního stavu v porovnání s pacienty na placebu hodnocené podle Health Assessment Questionnaire‑Disability Index (HAQ‑DI) v týdnu 24 a 16, v uvedeném pořadí. Zlepšení HAQ‑DI skóre bylo pozorováno bez ohledu na předchozí expozici anti‑TNFα. Podobná odpověď byla pozorována v PsA studii 1.Pacienti léčení sekukinumabem hlásili významné zlepšení se zdravím související kvality života měřené pomocí Short Form-36 Health Survey Physical Component Summary (SF‑36 PCS) skóre (p<0,001). Prokázalo se též statisticky významé zlepšení, což bylo demonstrováno ve výzkumných cílech hodnocených Functional Assessment of Chronic Illness Therapy – Fatigue (FACIT-F) skóre u dávky 150 mg a 300 mg v porovnání s placebem (7,97, respektive 5,97 versus 1,63) a tato zlepšení byla zachována až do týdne 104 v PsA studii 2.
Podobné odpovědi byly pozorovány v PsA studii 1 a účinnost přetrvávala do týdne 52.
Axiální spondylartritida (axSpA
Ankylozující spondylitida (AS) / radiografická axiální spondylartritida
Bezpečnost a účinnost sekukinumabu byla hodnocena u 816 pacientů ve třech randomizovaných, dvojitě zaslepených, placebem kontrolovaných studiích fáze III u pacientů s aktivní ankylozující spondylitidou (AS) s Bath Ankylosing Spondylitis Disease Activity indexem (BASDAI) ≥4 přes léčbu nesteroidními protizánětlivými léky (NSAID), kortikosteroidy nebo chorobu modifikujícími antirevmatiky (DMARD). Pacienti ve studii ankylozující spondylitidy 1 (AS studie 1) a studii ankylozující spondylitidy 2 (AS studie 2) měli stanovenu diagnózu AS s mediánem 2,7 až 5,8 roku. V obou studiích bylo primárním cílem nejméně 20% zlepšení Assessment of SpondyloArthritis International Society kritéria (ASAS 20) v týdnu 16.Ve studii ankylozující spondylitidy 1 (AS studie 1), studii ankylozující spondylitidy 2 (AS studie 2) a studii ankylozující spondylitidy 3 (AS studie 3) 27,0 %, respektive 38,8 % a 23,5 % pacientů, bylo dříve léčeno anti-TNFα přípravkem a léčba anti-TNFα přípravkem byla ukončena buď pro nedostatek účinku nebo nesnášenlivost (anti-TNFα-IR pacienti).
AS Studie 1 (MEASURE 1) hodnotila 371 pacientů, z nichž užívalo současně 14,8 %, respektive 33,4 %, MTX nebo sulfasalazin. Pacienti randomizovaní na sekukinumab dostávali dávku 10 mg/kg intravenózně v týdnech 0, 2 a 4, následovanou buď 75 mg nebo 150 mg subkutánně každý měsíc počínaje týdnem 8. Pacienti randomizovaní na placebo byli převedeni na sekukinumab (buď 75 mg nebo 150 mg subkutánně) s následnou stejnou dávkou každý měsíc, non respondéři v 16. týdnu (časná záchrana) a všichni ostatní ve 24. týdnu.
AS studie 2 (MEASURE 2) hodnotila 219 pacientů, z nichž užívalo současně 11,9 %, respektive 14,2 %, MTX nebo sulfasalazin. Pacienti randomizovaní na sekukinumab dostávali dávku 75 mg nebo 150 mg subkutánně v týdnech 0, 1, 2, 3 a 4, s následnou stejnou dávkou každý měsíc. V týdnu 16 byli pacienti při zahájení randomizovaní na placebo re-randomizováni na sekukinumab (buď 75 mg nebo 150 mg subkutánně) každý měsíc.
AS Studie 3 (MEASURE 3) hodnotila 226 pacientů, z nichž užívalo současně 13,3 %, respektive 23,5 %, MTX nebo sulfasalazin. Pacienti randomizovaní na sekukinumab dostávali dávku 10 mg/kg intravenózně v týdnech 0, 2 a 4, následovanou subkutánním podáním buď 150mg nebo 300mg každý měsíc. V týdnu 16 byli pacienti při zahájení randomizovaní na placebo re-randomizováni na sekukinumab (buď 150 mg nebo 300 mg subkutánně) každý měsíc. Primárním cílem bylo ASAS 20 v týdnu 16. Pacienti byli zaslepeni co se týče léčebného režimu do týdne 52 a studie pokračovala do týdne 152.
Známky a příznaky:
V AS studii 2 znamenala léčba 150 mg sekukinumabu v porovnání s placebem větší zlepšení v hodnocení aktivity choroby v týdnu 16 (viz tabulka 12).Tabulka 12 Klinická odpověď v AS studii 2 v týdnu 16
Výsledek (p-hodnota versus placebo)
Placebo
(n = 74)75 mg
(n = 73)150 mg
(n = 72)ASAS 20 odpověď, %
28,4
41,1
61,1***
ASAS 40 20 odpověď, %
10,8
26,0
36,1***
hsCRP, (post‑BSL/BSL ratio)
1,13
0,61
0,55***
ASAS 5/6, %
8,1
34,2
43,1***
ASAS částečná remise, %
4,1
15,1
13,9
BASDAI 50, %
10,8
24,7*
30,6**
ASDAS-CRP velké zlepšení
4,1
15,1*
25,0***
* p<0,05, ** p<0,01, *** p<0,001; versus placebo
Všechny p-hodnoty jsou nastaveny na mnohočetné testování založené na předem definované hierarchii, s výjimkou BASDAI 50 a ASDAS-CRP
Pacienti s chybějícími údaji o léčebné odpovědi byli považováni za non-respondéry.ASAS: Assessment of SpondyloArthritis International Society Criteria; BASDAI: Bath Ankylosing Spondylitis Disease Activity Index; hsCRP: high‑sensitivity C‑reactive protein; ASDAS: Ankylosing Spondylitis Disease Activity Score; BSL: baseline
V AS studii 2 se nástup účinku 150 mg sekukinumabu projevil již v týdnu 1 (superiorita Cosentyxu vůči placebu u odpovědi ASAS 20) a v týdnu 2 (superiorita Cosentyxu vůči placebu u odpovědi ASAS 40).
ASAS 20 se zlepšila v týdnu 16 jak u anti‑TNFα-naivních pacientů (68,2 % versus 31,1 %; p<0,05), tak u anti‑TNFα‑IR pacientů (50,0 % versus 24,1 %; p<0,05) u 150 mg sekukinumabu v porovnání s placebem.
V AS studii 1 a AS studii 2 došlo u pacientů léčených sekukinumabem (150 mg v AS Studii 2 a oběma režimy AS studie 1) v 16. týdnu k významnému zlepšení známek a příznaků se srovnatelným rozsahem odpovědi. Účinnost přetrvávala do 52. týdne jak u anti-TNFα-naivních, tak u anti-TNFα-IR pacientů. V AS studii 2 bylo v 52. týdnu stále léčeno 61 pacientů (84,7 %) ze 72 pacientů iniciálně randomizovaných na 150 mg sekukinumabu. Ze 72 pacientů randomizovaných na 150 mg sekukinumabu, 45, respektive 35, vykázalo ASAS 20/40 odpověď.
V AS studii 3, došlo u pacientů léčených sekukinumabem (150 mg a 300 mg) ke zlepšení známek a příznaků a vykazovali, nezávisle na dávce, srovnatelnou odpověď na léčbu co se týká účinnosti, která byla superiorní vůči placebu v týdnu 16 pro primární cílový parametr (ASAS 20). Celkově byla míra odezvy účinnosti u skupiny 300 mg trvale vyšší ve srovnání se skupinou 150 mg pro sekundární cílové parametry. Během zaslepeného období byly odpovědi ASAS 20 a ASAS 40 69,7% a 47,6% pro 150 mg a 74,3% a 57,4% pro 300 mg v 52. týdnu. Reakce ASAS 20 a ASAS 40 přetrvávaly až do týdne 156 (69,5% a 47,6% pro 150 mg oproti 74,8% a 55,6% pro 300 mg). Vyšší míra odpovědi ve prospěch 300 mg byla také pozorována u odpovědi na ASAS parciální remise (ASAS PR) v 16. týdnu a přetrvávala až do týdne 156. U pacientů s anti-TNFα-IR (n=36) byly pozorovány větší rozdíly v míře odezvy, preferující dávku 300 mg nad 150 mg, ve srovnání s anti-TNFα-naivními pacienty (n=114).
Mobilita páteře
Pacienti léčení sekukinumabem v dávce 150 mg vykázali zlepšenou mobilitu páteře hodnocenou jako změnu BASMI v týdnu 16 v porovnání s hodnotami před léčbou v AS studii 1 (‑0,40 versus ‑0,12 pro placebo; p=0,0114) a v AS studii 2 (‑0,51 versus ‑0,22 pro placebo; p=0,0533). Tato zlepšení přetrvávala do týdne 52.Funkční stav a se zdravím související kvalita života
V AS studii 1 a studii 2, vykázali pacienti léčení 150 mg sekukinumabu zlepšení zdravotního stavu a kvality života, což bylo měřeno AS Quality of Life Dotazníkem (ASQoL) (p=0,001) a SF-36 Physical Component Summary (SF-36PCS) (p<0,001). Pacienti léčení 150 mg sekukinumabu také vykázali statisticky významné zlepšení ve fyzické funkci ve výzkumných cílech, což bylo zhodnoceno Bath Ankylosing Spondylitis Functional Indexem (BASFI) v porovnání s placebem (-2,15 versus -0,68), a v únavě, což bylo zhodnoceno Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-Fatigue) škálou v porovnání s placebem (8,10 versus 3,30). Tato zlepšení přetrvávala do týdne 52.Non-radiografická axiální spondylartritida (nr-axSpA)
Bezpečnost a účinnost sekukinumabu byla hodnocena u 555 pacientů v jedné randomizované dvojitě slepé placebem kontrolované studii fáze III (PREVENT), sestávající z 2leté základní fáze a 2leté prodloužené fáze, u pacientů s aktivní non-radiografickou axiální spondylartritidou (nr-axSpA) splňující klasifikační kritéria Assessment of SpondyloArthritis International Society (ASAS) pro axiální spondylartritidu (axSpA) bez radiografického průkazu změn v sakroiliakálních kloubech, které by splňovaly modifikovaná New York kritéria pro ankylozující spondylitidu (AS). Zařazení pacienti měli aktivní onemocnění, definované jako Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4, a hodnotu na vizuální analogové škále (VAS) pro celkovou bolest v zádech ≥40 (na stupnici 0-100 mm), navzdory současné nebo předchozí terapii nesteroidními protizánětlivými léčivy (NSAID) a zvýšený C-reaktivní protein (CRP) a/nebo známky sakroiliitidy při zobrazení magnetickou rezonancí (MRI). Pacienti v této studii měli diagnózu axSpA v průměru 2,1 až 3,0 roku a 54% účastníků studie byly ženy.Ve studii PREVENT bylo 9,7% pacientů léčeno nejdříve anti-TNFα přípravkem a anti-TNFα přípravek byl vysazen z důvodu nedostatečné účinnosti nebo nesnášenlivosti (anti-TNFα-IR pacienti).
Ve studii PREVENT užívalo 9,9% a 14,8% pacientů souběžně MTX nebo sulfasalazin. Ve dvojitě zaslepeném období pacienti dostávali buď placebo nebo sekukinumab po dobu 52 týdnů. Pacienti randomizovaní na sekukinumab používali 150 mg subkutánně v týdnech 0, 1, 2, 3 a 4, po nichž následovala stejná dávka každý měsíc, nebo jednou měsíčně injekce se 150 mg sekukinumabu. Primárním cílovým parametrem bylo alespoň 40% zlepšení kritéria Assessment of SpondyloArthritis International Society (ASAS 40) v 16. týdnu u anti-TNFα-naivních pacientů.
Příznaky a symptomy:
Ve studii PREVENT vedla léčba sekukinumabem 150 mg k významnému zlepšení míry aktivity choroby v porovnání s placebem v 16. týdnu. Tato míra aktivity zahrnuje ASAS 40, ASAS 5/6, skóre BASDAI, BASDAI 50, CRP s vysokou citlivostí (hsCRP), ASAS 20 a částečnou remisi ASAS v porovnání s placebem (tabulka 13). Odpověď přetrvávala až do 52. týdne.Tabulka 13 Klinická odpověď ve studii PREVENT v týdnu 16
Výsledek (p-hodnota versus placebo)
Placebo
150 mg1
Počet anti-TNFα-naivních randomizovaných pacientů
171
164
Odpověď ASAS 40, %
29,2
41,5*
Celkový počet randomizovaných pacientů
186
185
Odpověď ASAS 40, %
28,0
40,0*
ASAS 5/6, %
23,7
40,0*
BASDAI, LS průměrná změna od výchozí hodnoty skóre
-1,46
-2,35*
BASDAI 50, %
21,0
37,3*
hsCRP, (poměr post-BSL/BSL)
0,91
0,64*
Odpověď ASAS 20, %
45,7
56,8*
Částečná remise ASAS, %
7,0
21,6*
*p<0,05 versus placebo
Všechny p-hodnoty byly upraveny pro větší počet testů na základě předem definované hierarchie
Pro chybějící binární cíl byla použita imputace non-respondérů
1Sekukinumab 150 mg s.c. v týdnech 0, 1, 2, 3 a 4 následovaný stejnou dávkou každý měsícASAS: Assessment of SpondyloArthritis International Society Criteria; BASDAI: Bath Ankylosing Spondylitis Disease Activity Index; hsCRP: C-reaktivní protein s vysokou citlivostí; BSL: výchozí hodnota; LS: Nejmenší čtverce
Nástup účinku sekukinumabu 150 mg ve studii PREVENT pro ASAS 40 u anti-TNFα-naivních pacientů (superiorní vůči placebu) se objevil již ve 3. týdnu. Procento ze všech anti-TNFα-naivních pacientů, kteří dosáhli odpovědi ASAS 40 v závislosti na čase je uvedeno na obrázku 3.
Obrázek 3 ASAS 40 odpovědi u anti-TNFα naivních pacientů ve studii PREVENT v průběhu času do 16. týdne
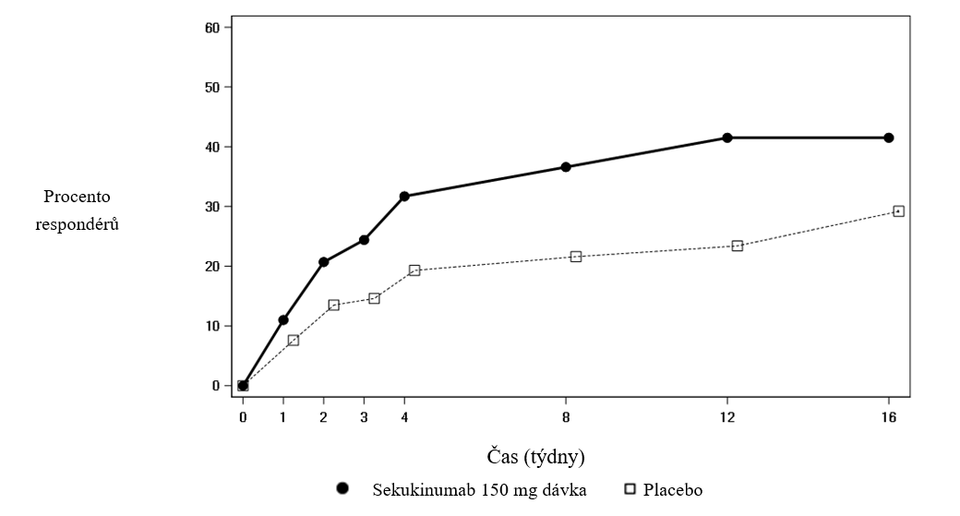
V 16. týdnu se u sekukinumabu 150 mg v porovnání s placebem objevilo u anti-TNFα-IR pacientů zlepšení ASAS 40 odpovědi.
Fyzické funkce a se zdravím související kvalita života:
Pacienti léčeni sekukinumabem 150 mg vykazovali statisticky významná zlepšení fyzické funkce do 16. týdne ve srovnání s pacienty léčenými placebem, hodnocené podle BASFI (týden 16: -1,75 versus -1,01, p<0,05). Pacienti léčeni sekukinumabem zaznamenali významné zlepšení ve srovnání s pacienty na placebu do 16. týdne v kvalitě života související se zdravím hodnocené podle ASQoL (průměrná změna LS: 16. týden: -3,45 versus -1,84, p<0,05) a SF-36 Physical Component Summary (SF-36 PCS) (LS průměrná změna: týden 16: 5,71 versus 2,93, p<0,05). Tato zlepšení přetrvávala až do 52. týdne.Spinální mobilita:
Spinální mobilita byla hodnocena podle BASMI až do 16. týdne. Numericky větší zlepšení byla prokázána u pacientů léčených sekukinumabem v porovnání s pacienty na placebu ve 4., 8., 12. a 16. týdnu.Inhibice zánětu při zobrazení magnetickou rezonancí (MRI):
Příznaky zánětu byly hodnoceny pomocí MRI na začátku a v 16. týdnu a byly vyjádřeny jako změna oproti základní hodnotě v Berlin SI-joint oedema skóre pro sakroiliakální klouby a ASspiMRI-a skóre a Berlin spine skóre pro páteř. U pacientů léčených sekukinumabem byla pozorována inhibice zánětlivých příznaků jak pro sakroiliakální klouby, tak i pro páteř. Průměrná změna oproti výchozím hodnotám v Berlin SI-joint oedema skóre byla -1,68 u pacientů léčených sekukinumabem 150 mg (n=180) oproti -0,39 u pacientů na placebu (n=174) (p<0,05). - sekukinumab 300 mg v týdnech 0, 1, 2, 3 a 4 následovaný stejnou dávkou každé 2 týdny
-
Pediatrická populace
Ložisková psoriáza u pediatrické populace
Bylo prokázáno, že sekukinumab zlepšuje známky a příznaky a kvalitu života u pediatrických pacientů s ložiskovou psoriázou ve věku 6 let a starších (viz tabulky 15 a 17).Těžká ložisková psoriáza
Bezpečnost a účinnost sekukinumabu byla hodnocena v randomizované, dvojitě zaslepené, placebem a etanerceptem kontrolované studii fáze III u dětských pacientů ve věku od 6 do <18 let s těžkou ložiskovou psoriázou, definovanou pomocí skóre PASI ≥20, IGA mod 2011 skóre 4 a poměru BSA ≥ 10 %, kteří byli kandidáty na systémovou terapii. Přibližně 43 % pacientů mělo předchozí expozici fototerapii, 53 % konvenční systémové terapii, 3 % biologické léčbě a 9 % mělo souběžnou psoriatickou artritidu.V pediatrické studii psoriázy 1 bylo hodnoceno 162 pacientů, kteří byli randomizováni k podávání nízké dávky sekukinumabu (75 mg pro tělesnou hmotnost <50 kg nebo 150 mg pro tělesnou hmotnost ≥50 kg), vysoké dávky sekukinumabu (75 mg pro tělesnou hmotnost <25 kg, 150 mg pro tělesnou hmotnost mezi >25 kg a <50 kg nebo 300 mg pro tělesnou hmotnost >50 kg), nebo placeba v týdnech 0, 1, 2, 3 a 4 následované stejnou dávkou každé 4 týdny, nebo etanerceptem. Pacienti randomizovaní na etanercept dostávali 0,8 mg/kg týdně (až do maxima 50 mg). Rozdělení pacientů podle hmotnosti a věku při randomizaci je popsáno v tabulce 14.
Tabulka 14 Rozdělení pacientů podle hmotnosti a věku v pediatrické studii psoriázy 1
Randomizační vrstvy Popis Sekukinumab
nízká dávka
n=40Sekukinumab
vysoká dávka
n=40Placebo
n=41Etanercept
n=41Celkem
N=162Věk 6-<12 let 8 9 10 10 37 ≥12-<18 let 32 31 31 31 125 Hmotnost <25 kg 2 3 3 4 12 ≥25-<50 kg 17 15 17 16 65 ≥50 kg 21 22 21 21 85 Pacienti randomizovaní na placebo a bez odpovědi na léčbu ve 12. týdnu byli přeřazeni do skupiny s nízkou nebo vysokou dávkou sekukinumabu (dávka podle skupiny tělesné hmotnosti) a dostávali studijní medikaci ve 12., 13., 14. a 15. týdnu, následovanou stejnými dávkami každé 4 týdny počínaje 16. týdnem. Ko-primárními cílovými parametry byly podíl pacientů, kteří dosáhli odpovědi PASI 75 a odpovědi IGA mod 2011 „čistá“ nebo „téměř čistá“ (0 nebo 1) ve 12. týdnu.
Během 12týdenního placebem kontrolovaného období byla účinnost jak nízké, tak vysoké dávky sekukinumabu srovnatelná pro ko-primární cílové parametry. Odhady poměru šancí ve prospěch obou dávek sekukinumabu byly statisticky významné pro odpovědi PASI 75 a IGA mod 2011 0 nebo 1.
Během 52 týdnů po první dávce byli všichni pacienti sledováni z hlediska účinnosti a bezpečnosti. Podíl pacientů, kteří dosáhli odpovědí PASI 75 a IGA mod 2011 „čistá“ nebo „téměř čistá“ (0 nebo 1) vykazoval rozdíl mezi skupinou léčenou sekukinumabem a placebem při první návštěvě po zahájení léčby ve 4. týdnu, přičemž rozdíl se stal výraznějším ve 12. týdnu. Odpověď přetrvávala po celou dobu 52 týdnů (viz tabulka 15). V průběhu 52týdenního časového období také přetrvávalo zlepšení v počtu odpovědí PASI 50, 90, 100 a Children’s Dermatology Life Quality Index (CDLQI) skóre 0 nebo 1.
Kromě toho byl počet odpovědi PASI 75, IGA 0 nebo 1, PASI 90 ve 12. a 52. týdnu ve skupinách se sekukinumabem s nízkou i vysokou dávkou vyšší než u pacientů léčených etanerceptem (viz tabulka 15).
Po 12. týdnu byla účinnost jak nízké, tak vysoké dávky sekukinumabu srovnatelná, i když účinnost vysoké dávky byla vyšší u pacientů ≥ 50 kg. Bezpečnostní profily nízké dávky a vysoké dávky byly srovnatelné a konzistentní s bezpečnostním profilem u dospělých.
Tabulka 15 Přehled klinické odpovědi u těžké pediatrické psoriázy v týdnech 12 a 52 (pediatrická studie psoriázy 1)*
Kritérium odpovědi Srovnání odpovědi 'test' 'kontrola' Odhad poměru šancí 'test' vs. 'kontrola' n**/m (%) n**/m (%) (95% CI) p-hodnota Týden 12*** PASI 75 sekukinumab nízká dávka vs. placebo 32/40 (80,0) 6/41 (14,6) 25,78 (7,08; 114,66) <0,0001 sekukinumab vysoká dávka vs. placebo 31/40 (77,5) 6/41 (14,6) 22,65 (6,31; 98,93) <0,0001 sekukinumab nízká dávka vs. etanercept 32/40 (80,0) 26/41 (63,4) 2,25 (0,73; 7,38) sekukinumab vysoká dávka vs. etanercept 31/40 (77,5) 26/41 (63,4) 1,92 (0,64; 6,07) IGA 0/1 sekukinumab nízká dávka vs. placebo 28/40 (70,0) 2/41 (4,9) 51,77 (10,02; 538,64) <0,0001 sekukinumab vysoká dávka vs. placebo 24/40 (60,0) 2/41 (4,9) 32,52 (6,48; 329,52) <0,0001 sekukinumab nízká dávka vs. etanercept 28/40 (70,0) 14/41 (34,1) 4,49 (1,60; 13,42) sekukinumab vysoká dávka vs. etanercept 24/40 (60,0) 14/41 (34,1) 2,86 (1,05; 8,13) PASI 90 sekukinumab nízká dávka vs. placebo 29/40 (72,5) 1/41 (2,4) 133,67 (16,83; 6395,22) <0,0001 sekukinumab vysoká dávka vs. placebo 27/40 (67,5) 1/41 (2,4) 102,86 (13,22; 4850,13) <0,0001 sekukinumab nízká dávka vs. etanercept 29/40 (72,5) 12/41 (29,3) 7,03 (2,34; 23,19) sekukinumab vysoká dávka vs. etanercept 27/40 (67,5) 12/41 (29,3) 5,32 (1,82; 16,75) Týden 52 PASI 75 sekukinumab nízká dávka vs. etanercept 35/40 (87.5) 28/41 (68,3) 3,12 (0,91; 12,52) sekukinumab vysoká dávka vs. etanercept 35/40 (87.5) 28/41 (68,3) 3,09 (0,90; 12,39) IGA 0/1 sekukinumab nízká dávka vs. etanercept 29/40 (72.5) 23/41 (56,1) 2,02 (0,73; 5,77) sekukinumab vysoká dávka vs. etanercept 30/40 (75.0) 23/41 (56,1) 2,26 (0,81; 6,62) PASI 90 sekukinumab nízká dávka vs. etanercept 30/40 (75.0) 21/41 (51,2) 2,85 (1,02; 8,38) sekukinumab vysoká dávka vs. etanercept 32/40 (80.0) 21/41 (51,2) 3,69 (1,27; 11,61) * u chybějících hodnot non-respondérů byla použita imputace
** n je počet responderů, m = počet vyhodnotitelných pacientů
*** rozšířené okno pro návštěvu v 12. týdnu
Poměr šancí, 95% interval spolehlivosti, a p-hodnota pochází z exaktního logistického regresního modelu s léčebnou skupinou, s kategoriemi tělesná hmotnost před léčbou a věk jako faktoryVyšší podíl pediatrických pacientů léčených sekukinumabem uváděl zlepšení se zdravím související kvality života, měřeno pomocí skóre CDLQI 0 nebo 1 v porovnání s placebem ve 12. týdnu (nízká dávka 44,7 %, vysoká dávka 50 %, placebo 15 %). Během celého průběhu do a včetně týdne 52 byly obě skupiny s dávkou sekukinumabu numericky větší než skupina s etanerceptem (nízká dávka 60,6 %, vysoká dávka 66,7 %, etanercept 44,4 %).
Středně těžká až těžká ložisková psoriáza
U sekukinumabu se předpokládala účinnost při léčbě pediatrických pacientů se středně těžkou ložiskovou psoriázou na základě prokázaného vztahu účinnosti a odpovědi na expozici u dospělých pacientů se středně těžkou až těžkou ložiskovou psoriázou a podobnosti průběhu onemocnění, patofyziologie a účinku léčiva u dospělých a pediatrických pacientů při stejných úrovních expozice.Kromě toho byla bezpečnost a účinnost sekukinumabu hodnocena v otevřené dvouramenné multicentrické studii fáze III s paralelní skupinou u dětských pacientů ve věku od 6 do <18 let se střední až těžkou ložiskovou psoriázou, definovanou skóre PASI ≥12, skóre IGA mod 2011 ≥3 a poměr BSA>10 %, kteří byli kandidáty na systémovou terapii.
V pediatrické studii psoriázy 2 bylo hodnoceno 84 pacientů, kteří byli randomizováni k podávání nízké dávky sekukinumabu (75 mg u tělesné hmotnosti <50 kg nebo 150 mg u tělesné hmotnosti ≥ 50 kg) nebo vysoké dávky sekukinumabu (75 mg pro tělesnou hmotnost <25 kg, 150 mg pro tělesnou hmotnost mezi >25 kg a <50 kg nebo 300 mg pro tělesnou hmotnost>50 kg) v týdnech 0, 1, 2, 3 a 4, následovaná stejnou dávkou každé 4 týdny. Rozdělení pacientů podle hmotnosti a věku při randomizaci je popsáno v tabulce 16.
Tabulka 16 Rozdělení pacientů podle hmotnosti a věku v pediatrické studii psoriázy 2
Podskupiny Popis Sekukinumab
Nízká dávka
n=42Sekukinumab
Vysoká dávka
n=42Celkem
N=84Věk 6-<12 let 17 16 33 ≥12-<18 let 25 26 51 Hmotnost <25 kg 4 4 8 ≥25-<50 kg 13 12 25 ≥50 kg 25 26 51 Ko-primárními cílovými parametry byly podíl pacientů, kteří dosáhli odpovědi PASI 75 a odpovědi IGA mod 2011 „čistá“ nebo „téměř čistá“ (0 nebo 1) ve 12. týdnu.
Účinnost nízké i vysoké dávky sekukinumabu byla srovnatelná a vykazovala statisticky významné zlepšení ve srovnání s anamnézou placeba pro ko-primární cílové parametry. Odhadovaná pozdější pravděpodobnost pozitivního léčebného účinku byla 100 %.
Účinnost byla u pacientů sledována po dobu 52 týdnů po prvním podání. Účinnost (definovaná jako odpověď PASI 75 a IGA mod 2011 „čistá“ nebo „téměř čistá“ [0 nebo 1]) byla pozorována již při první návštěvě po základní návštěvě ve 2. týdnu a podíly pacientů, kteří dosáhli odpovědi PASI 75 a IGA mod 2011 „čistá“ nebo „téměř čistá“ (0 nebo 1) vzrostly až do 24. týdne a byly udržovány až do 52. týdne. Zlepšení v PASI 90 a PASI 100 bylo také pozorováno ve 12. týdnu a zvýšeno až do 24. týdne a bylo udržováno do 52. týdne (viz tabulka 17).
Bezpečnostní profily nízké dávky a vysoké dávky byly srovnatelné a konzistentní s bezpečnostním profilem u dospělých.
Tabulka 17 Přehled klinické odpovědi u středně těžké až těžké pediatrické psoriázy v týdnech 12 a 52 (pediatrická studie psoriázy 2)*
Týden 12 Týden 52 nízká dávka vysoká dávka nízká dávka vysoká dávka Počet pacientů 42 42 42 42 Odpověď PASI 75n (%) 39 (92,9 %) 39 (92,9 %) 37 (88,1 %) 38 (90,5 %) Odpověď IGA mod 2011 „čistá“ nebo „téměř čistá“ n (%) 33 (78,6 %) 35 (83,3 %) 36 (85,7 %) 35 (83,3 %) Odpověď PASI 90n (%) 29 (69 %) 32 (76,2 %) 32 (76,2 %) 35 (83,3 %) Odpověď PASI 100n (%) 25 (59,5 %) 23 (54,8 %) 22 (52,4 %) 29 (69,0 %) * u chybějících hodnot non-respondérů byla použita imputace Tyto výsledky u pediatrické populace se středně těžkou až těžkou ložiskovou psoriázou potvrdily výše uvedené prediktivní předpoklady na základě vztahu účinnosti a expozice u dospělých pacientů.
Ve skupině s nízkou dávkou dosáhlo 50 % a 70,7 % pacientů skóre CDLQI 0 nebo 1 ve 12. a 52. týdnu. Ve skupině s vysokou dávkou dosáhlo 61,9 % a 70,3 % skóre CDLQI 0 nebo 1 ve 12. a 52. týdnu.
Juvenilní idiopatická artritida (JIA)
Artritida související s entezitidou (ERA) a juvenilní psoriatická artritida (JPsA)
Bezpečnost a účinnost sekukinumabu byly hodnoceny u 86 pacientů ve 3 částech dvojitě zaslepené, placebem kontrolované, událostmi řízené (event-driven), randomizované studie fáze III u pacientů ve věku od 2 do <18 let s aktivní ERA nebo JPsA, kteří byli diagnostikováni podle modifikovaných JIA klasifikačních kritérií Mezinárodní ligy asociací pro revmatologii (International League of Associations for Rheumatology, ILAR). Studie se skládala z otevřené fáze (část 1), ve které všichni pacienti dostávali sekukinumab do 12. týdne. Pacienti, kteří ve 12. týdnu dosáhli odpovědi JIA ACR30, vstoupili do dvojitě zaslepené fáze (část 2), kde byli randomizováni v poměru 1:1, aby pokračovali v léčbě sekukinumabem nebo byli převedeni na placebo (randomizované vysazení) do 104. týdne nebo do výskytu příznaků vzplanutí. Pacienti se vzplanutím onemocnění vstoupili do otevřené studie, ve které byli léčeni sekukinumabem do 104. týdne (část 3).Při zařazení do studie měli pacienti s JIA v 60,5 % podtyp ERA a 39,5 % podtyp JPsA a vykazovali nedostatečnou odpověď nebo intoleranci na ≥1 chorobu modifikující antirevmatika (DMARD) a ≥1 nesteroidní antiflogistika (NSAID). Při zahájení studie bylo hlášeno užívání MTX u 65,1 % pacientů; (63,5 % [33/52] pacientů s ERA a 67,6 % [23/34] pacientů s JPsA). Dvanáct z 52 pacientů s ERA současně užívalo sulfasalazin (23,1 %). Pacienti s tělesnou hmotností <50 kg (n=30) při zahájení studie dostávali dávku 75 mg a pacienti s tělesnou hmotností ≥50 kg (n=56) dostávali dávku 150 mg. Věkové rozmezí při zahájení studie bylo od 2 do 17 let, 3 pacienti byli ve věku 2 až <6 let, 22 pacientů ve věku 6 až <12 let a 61 pacientů ve věku 12 až <18 let. Skóre JADAS-27 bylo při zahájení studie 15,1 (SD: 7,1).
Primárním cílovým parametrem byla doba do vzplanutí onemocnění ve fázi randomizovaného vysazení (část 2). Vzplanutí onemocnění bylo definováno jako ≥30% zhoršení odpovědi JIA ACR u alespoň tří ze šesti kritérií a ≥30% zlepšení odpovědi JIA ACR u ne více jak jednoho ze šesti kritérií a minimálně dvou aktivních kloubů.
Na konci části 1 dosáhlo 75 pacientů z 86 (87,2 %) odpovědi JIA ACR30 a vstoupilo do části 2.
Ve studii byl splněn primární cíl, protože bylo prokázáno statisticky významné prodloužení doby do vzplanutí onemocnění u pacientů léčených v části 2 sekukinumabem ve srovnání s placebem. U pacientů léčených v části 2 sekukinumabem bylo riziko vzplanutí sníženo o 72 % ve srovnání s pacienty, kterým bylo podáváno placebo (poměr rizika = 0,28; 95% CI: 0,13 až 0,63, p<0,001) (obrázek 4 a tabulka 18). Během části 2 došlo ke vzplanutí onemocnění celkem u 21 pacientů ve skupině s placebem (11 JPsA a 10 ERA) ve srovnání s 10 pacienty ve skupině se sekukinumabem (4 JPsA a 6 ERA).
Obrázek 4 Doba do vzplanutí onemocnění v části 2 podle metody Kaplana-Meiera
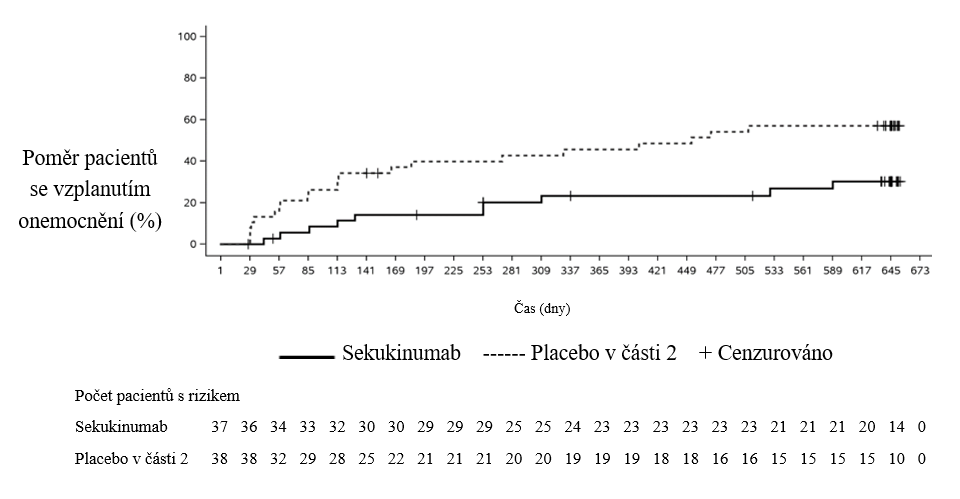
Tabulka 18 Analýza doby přežívání do vzplanutí onemocnění v části 2
(n=37) Placebo v části 2
(n=38)počet případů vzplanutí na konci části 2, n (%) 10 (27,0) 21 (55,3) Kaplanova-Meierova metoda: medián ve dnech (95% CI) NC (NC, NC) 453,0 (114,0; NC) poměr bez vzplanutí v 6. měsíci (95% CI) 85,8 (69,2; 93,8) 60.1 (42.7, 73.7) poměr bez vzplanutí ve 12. měsíci (95% CI) 76,7 (58,7; 87,6) 54,3 (37,1; 68,7) poměr bez vzplanutí v 18. měsíci (95% CI) 73,2 (54,6; 85,1) 42,9 (26,7; 58,1) poměr rizik k placebu: odhad (95% CI) 0,28 (0,13; 0,63) p‑hodnota ze stratifikovaného log‑rank testu <0,001** Analýza byla provedena u všech randomizovaných pacientů, kteří v části 2 dostali alespoň jednu dávku studijní medikace.
Sekukinumab: všichni pacienti, kteří nedostali žádné placebo. Placebo v části 2: všichni pacienti, kteří v části 2 užívali placebo a v další části/dalších částech užívali sekukinumab. NC = nelze spočítat. ** = Statisticky významná hodnota pro jednostrannou hladinu významnosti 0,025.V otevřené části 1 dostávali všichni pacienti sekukinumab do 12. týdne. Ve 12. týdnu dosáhlo 83,7 % dětí odpovědi JIA ACR50; 67,4 % odpovědi JIA ACR70 a 38,4 % odpovědi JIA ACR90 (obrázek 5). Nástup účinku sekukinumabu nastal již v 1. týdnu. Ve 12. týdnu bylo skóre JADAS-27 4,64 (SD:4,73) a průměrné snížení od výchozí hodnoty JADAS-27 bylo -10,487 (SD:7,23).
Obrázek 5 Odpověď JIA ACR30/50/70/90 u subjektů v části 1 do 12. týdne*
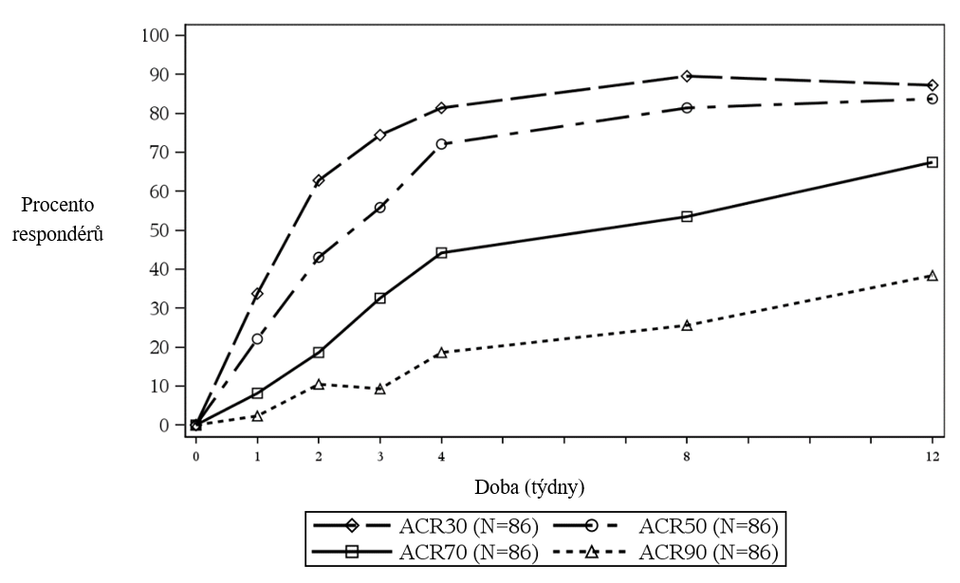
* u chybějících hodnot non-respondérů byla použita imputace
Údaje pro věkovou skupinu od 2 do <6 let jsou neprůkazné vzhledem k malému počtu pacientů mladších 6 let zahrnutých do studie.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Cosentyx u chronické idiopatické artritidy u dětských pacientů do 2 let (informace o použití u dětí viz bod 4.2).
-
6. FARMACEUTICKÉ ÚDAJE
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Irsko
8. REGISTRAČNÍ ČÍSLO/REGISTRAČNÍ ČÍSLA
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 15. ledna 2015
Datum posledního prodloužení registrace: 3. září 2019
10. DATUM REVIZE TEXTU
13. února 2025
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky https://www.ema.europa.eu.